
Whether you have been newly diagnosed with mast cell activation syndrome (MCAS) or if you have been struggling for years, you likely have been provided the same basic treatment options. Most patients are first offered Histamine 1 blockers (H1 blocker) which are antihistamines including Claritin, Allegra, Zyrtec, Xyzal, Benadryl and Hydroxyzine. Typically, a histamine 2 blocker is then added which is usually Pepcid now that Zantac is off of the market. If these medications at appropriate doses are insufficient, mast cell stabilizers such as cromolyn sodium and ketotifen (really both a mast cell stabilizer and an H1 blocker) are used. If prostaglandin D2 levels are high, aspirin therapy may be recommended and for those with leukotriene and asthma symptoms, Montelukast or zafirlukast can be implemented. If symptoms are very severe, steroids such as prednisone are offered. Long term immunotherapies such as Dupixent and Xolair are also considered. Unfortunately, while this may help some patients tolerate the world a bit better, it rarely eliminates the symptoms altogether.
After working with mast cell patients for so many years, it has become extremely clear to me that to begin to really improve patient outcome in those with MCAS, you have to find and then address the root cause. This is where things become a bit trickier as there is no “one-size-fits-all” root cause. At one point, we thought Ehlers-Danlos Syndrome (EDS) was a root cause. Now we know it is part of the triad: MCAS, EDS and dysautonomia. However, it still isn’t really a root cause. There are three things that I have found do drive mast cells to misbehave. This includes familial hypertryptasemia, immunodeficiency and neurological autoimmunity.
Familial Hypertryptasemia
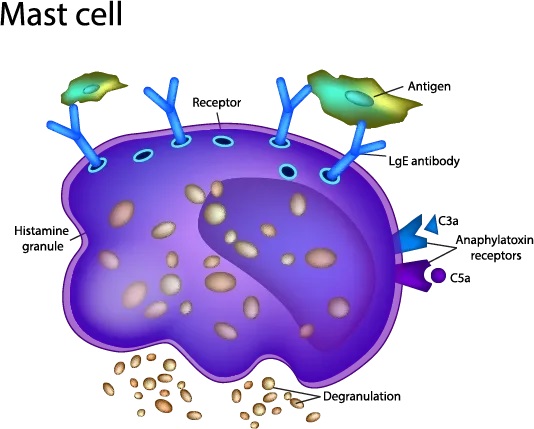
Related to a copy number variant (CNV—a fancy way of saying more than one copy of the gene) in the tryptase gene, familial hypertryptasemia also know has hereditary alpha tryptasemia is a genetic condition where there are multiple copies of the tryptase gene. Tryptase is released by the mast cell most commonly as a part of an anaphylactic response. It is also elevated in mastocytosis. In the case of this genetic condition, we find that patients present with symptoms including flushing, itching, swelling, hives, hypermobility/EDS, allergic reactions, food reactions, medication reactions, chemical sensitivities, IBS, dysmotility, acid reflux, dysautonomia, anxiety, depression and pain. No patient will have all of these symptoms, of course, but usually multiple symptoms will be present. The average tryptase level without this condition is typically between 3-5. Those with familial hypertryptasemia will have levels that are above 5 consistently. This can be well below an elevated tryptase level which is when levels increase above 11. Testing for the tryptase CNV is available at Gene by Gene.
Immunodeficiency
Immunodeficiency is another common driver of mast cell activation. This is because immunoglobulins are the “smart” part of our immune system that has memory. When we are exposed to a virus or bacteria that our body has been exposed to either by a vaccine or illness, it sends out immunoglobulins even more quickly to fight the infection than when the body was first exposed. When the body’s ability to produce immunoglobulins becomes compromised, then other portions of the immune system must step up to the plate so to speak and take over. Mast cells are part of the innate immune system. They are present since birth and amongst other things guard the periphery of our body (anywhere we come into contact with the outside world) from antigens. When things are working properly, part of this responsibility is calling in immunoglobulins for back up when they cannot do the job themselves. As a result of immunodeficiency (even subclinical immunodeficiency), we can find that the mast cells go onto high alert and are more likely to degranulate at the slightest trigger. This can then cause mast cell activation.
Immunoglobulin deficiencies can either be primary or secondary. Primary immunoglobulin deficiency includes disorders such as common variable immunodeficiency (CVID) and are genetic in nature. They are typically treated with IVIG and often prophylactic antibiotics. Secondary immunodeficiencies are acquired and not genetic. They tend to result when there are either chronic infectious agents, autoimmunity and/or aggressive treatment with antibiotics (such as what is often prescribed for Lyme disease). Secondary immunodeficiencies sometimes require IVIG. When levels are not low enough to warrant medical intervention, use of over-the-counter colostrum or bovine albumin immunoglobulins can be helpful.
For some patients, immunodeficiency may not be immunoglobulin based and may be more related to a mannose binding lectin deficiency (MBL deficiency). MBL is part of our complement system. This system is designed to help kill off pathogens. It may be triggered through one of three mechanisms: immunoglobulins, MBL or innate immune cells including mast cells. For this reason, when MBL is low it can also trigger mast cell activation. MBL deficiency is also genetic and may be caused by a heterozygous (one allele is affected) or homozygous (both alleles are affected) mutation. This is a milder immunodeficiency and often goes undiagnosed. Those affected often have more severe and frequent respiratory and gastrointestinal infections.
Neurological Autoimmunity
There also appears to be a strong overlap between MCAS and neurological autoimmunity. So many mast cell patients present with dysautonomia/POTS types of symptoms including tachycardia (rapid heart rate), low or high blood pressure, dizziness, nausea, gastroparesis, fainting, light sensitivity, sound sensitivity, etc. Many also present with neuropathy (nerve pain). Often these issues can be related to small fiber, large fiber and/or autonomic neuropathy. This happens often in conjunction with not just MCAS but with those with Ehlers-Danlos Syndrome as well.
Many of these patients can have an autoimmune disease known as autoimmune small fiber polyneuropathy. It can cause a variety of symptoms including those listed above. Further, many of these patients have neurological reactions that are easily confused for MCAS reactions and often become incredibly limited in terms of food tolerances. This disease is related to autoantibodies to either ganglioside and/or sulfatide. Traditional treatment may include the medication gabapentin which is helpful with nerve pain. Some of the more forward-thinking neurologists are using neurological doses of IVIG and are having extraordinary effects.